Chapter 9: Learning and Memory
9.6: Disorders of memory
Alzheimer’s disease
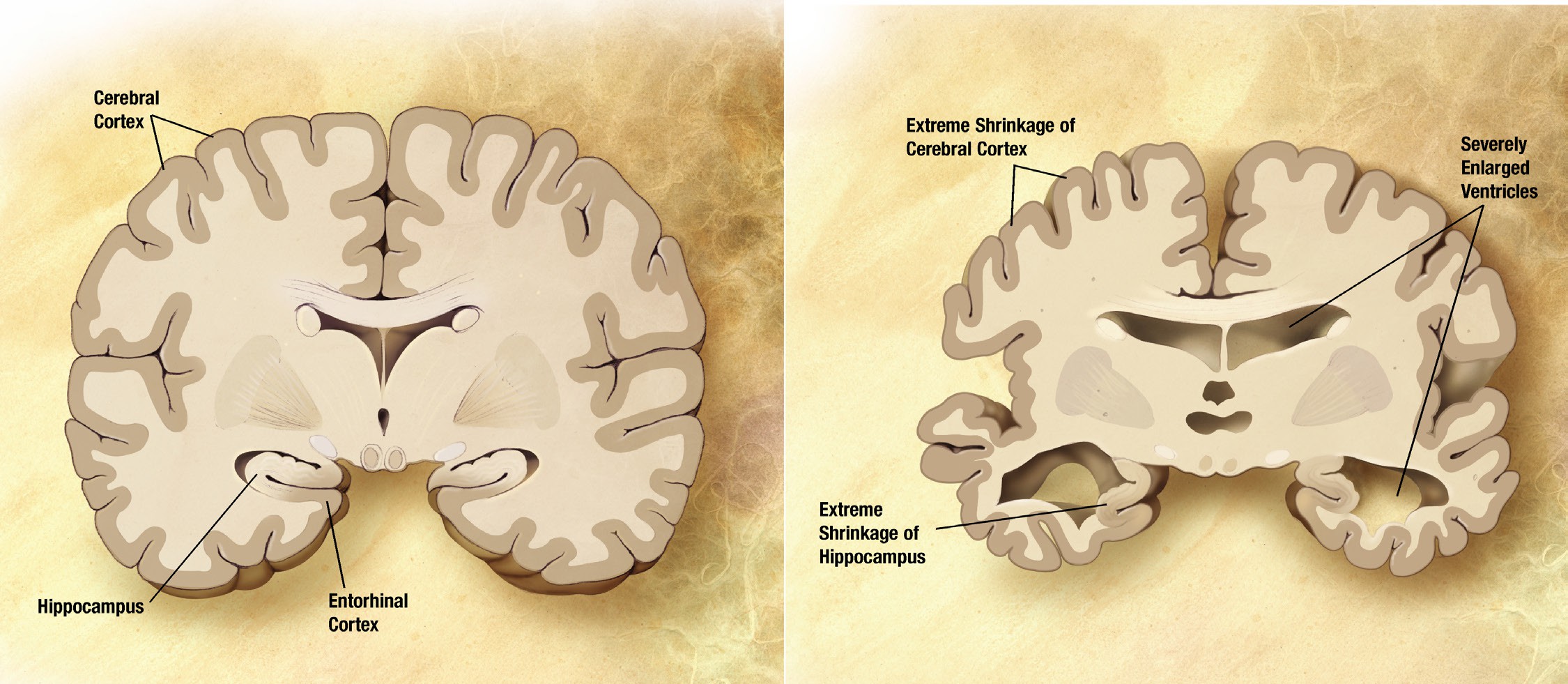
In 1907, Dr. Alois Alzheimer described one of his patients, a 50-year old woman named Auguste Deter, whose symptoms included profound cognitive impairment, memory deficits, and delusions. After Deter’s death, post-mortem analysis of her brain revealed anomalies: degenerating neurons that contained atypical tangles and deposits scattered between cells. These reports were the first of a disease we now call Alzheimer’s disease (AD). It is an irreversible, slowly progressing neurodegenerative condition that leads to deficits in thinking, behavior, and memory loss. Specifically, declarative memory is the first form of memory loss observed in AD patients. As the disease progresses, procedural memory loss becomes more apparent.
AD is a devastating disease that accounts for 60-80% of all cases of dementia and is the sixth leading cause of death in the United States. As for prevalence, approximately 10% of people older than 65 and nearly a third older of people older than 85 has AD.
AD is divided into two categories, familial and sporadic. Familial AD is diagnosed when a person is in their 50s or 60s, and is strongly influenced by genetic risk factors. This form of AD only makes up about 10% of cases. Sporadic AD is by far more common than familial AD, and is believed to be caused by a combination of old age and environmental factors in addition to genetic risk factors.
Several genetic risk factors are linked to AD risk. Apolipoprotein epsilon4 (ApoE4) is the greatest genetic risk factor identified, where individuals who are homozygous for the e4 polymorphism have a 12-times higher risk of developing AD than people with the more common e3 variant. Additionally, mutations in genes like amyloid-precursor protein (APP), presenilin-1 (PSEN1) presenilin-2 (PSEN2), and triggering receptors expressed on myeloid cells2 (TREM2) are all associated with higher risk of developing AD.
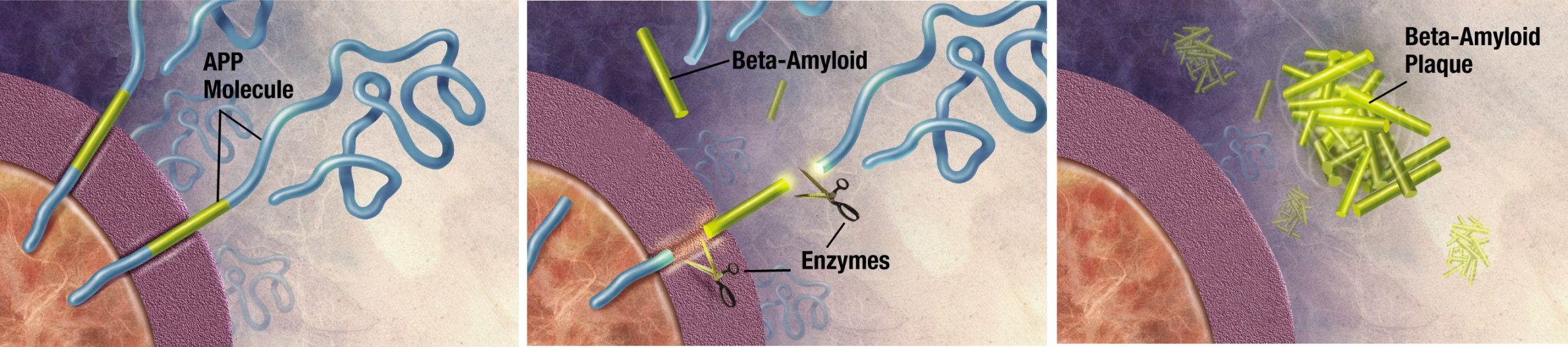
Pathologically, AD is characterized by the extracellular accumulation of amyloid- beta plaques (Aβ) and intracellular hyperphosphorylated neurofibrillary tau- tangles (NFT). The amyloid cascade hypothesis, originally proposed in 1992, has become the leading theory in the field describing how AD develops. The hypothesis suggests that the main driving factor of AD is the deposition of Aβ in the brain, and this in turn leads to neurodegeneration via cell death, abnormal protein buildup, and neuroinflammation. Aβ is produced from the cleavage of amyloid-precursor protein (APP), an integral membrane protein expressed by neurons. A class of secretase enzymes are responsible for the degradation of APP. When the alpha and gamma secretases degrade APP, the resulting Aβ protein is likely to clump together in unpredictable ways, leading to the formation of Aβ plaques (pronounced “A beta”). These plaques can cause neuronal death and lead to the cognitive deficits observed in AD patients. Two major genetic risk factors for AD, presenilin 1 and 2, are mutations of the gamma secretase that leads to increased APP cleavage.
Therapies are being developed to target and control the Aβ protein to mitigate AD symptoms. Recently an antibody-based drug has been approved by the FDA. This groundbreaking drug is unique from those traditionally prescribed because it directly targets Aβ.
However, the amyloid-cascade hypothesis does not tell the whole story. For one, there are patients who carry a heavy Aβ load but present no clinical symptoms ofAD. More so, in mouse models with APP mutations, they develop significant Aβ plaques, but have no accumulation of tau (see below) and no significant neurodegeneration. Additionally, there have been several drugs targeting Aβ that have failed in human trials.
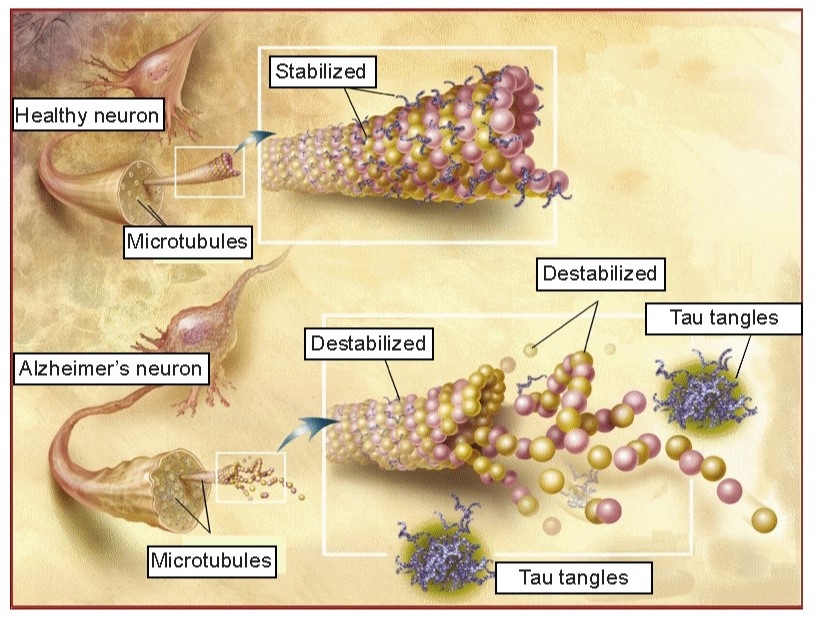
While these amyloid plaques seem to be the primary driving factor of AD, hyperphosphorylated tau and neurofibrillary tangles (NFTs) are also potential culprits in disease progression. NFTs are an intracellular pathological marker of AD and correlate strongly with cognitive deficits in AD. Tau protein is the major microtubule-associate protein (MAP) of mature neurons, and helps to function to maintain cellular morphology. Excess phosphorylation of this protein causes tau to accumulate inside the cell, leading to neuronal dysfunction and cell death. The presence of Aβ increases the levels of the tau, and vice versa, adding to the complexity and difficulty of treating AD. Tau pathology is also observed in other neurodegenerative diseases such as frontotemporal lobe dementia and Parkinson’s disease.
Although most discussion of AD revolves around plaques and tangles, it is well known that a host of pathological markers are also seen in AD, including neuroinflammation, oxidative stress, blood-brain barrier dysfunction, heavy metal dysregulation, mitochondrial impairment, and many more.
Korsakoff’s syndrome
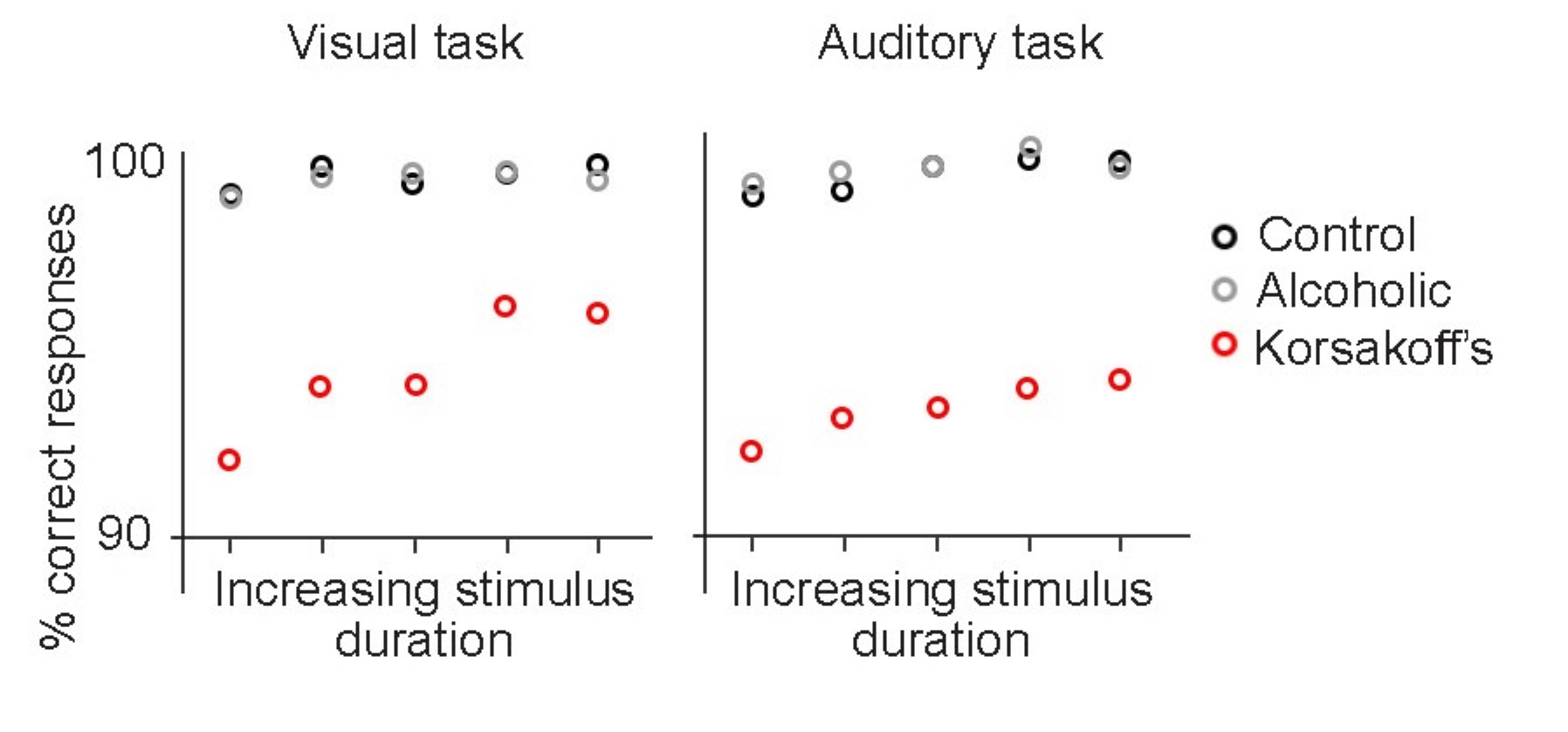
Korsakoff’s syndrome is a disorder resulting from a severe deficiency of thiamine, an essential vitamin that functions in metabolic processes. Dietary thiamine is found in whole grains, legumes such as beans and peas, as well as some meats and fishes. Healthy people with a well-balanced diet get sufficient thiamine, however gastrointestinal illnesses can cause an inability to absorb thiamine properly. Chronic alcohol misuse also impairs the body’s ability to take up thiamine, and is the most common cause of Korsakoff’s syndrome.
People with Korsakoff’s syndrome experience both retrograde and anterograde amnesia, as well as severely impaired short-term memory. The patients may experience a very strange behavior called confabulation, which is the fabrication of false memories ranging from subtle to wildly fantastical. People who confabulate do not consciously recognize that their statements are untrue, and are not intentionally trying to deceive others, which is why it is sometimes called “honest lying”. Generally, confabulation only happens as a person is trying to recall recent
autobiographical memories, while their semantic and procedural memories are less susceptible to confabulation. Scientists suggest that people confabulate as a compensatory mechanism to
make up for their retrograde amnesia. Destruction of both neurons and glia are seen in the brains of people with Korsakoff’s syndrome. As a result of this cell loss, there is often shrinkage of the cortex, thalamus, the hippocampus, cerebellum, and the mammillary bodies, paired structures located at the ventral surface of the brain close to the brain stem, themselves part of the limbic system.
Korsakoff’s syndrome can be treated by giving thiamine supplements and eliminating alcohol consumption. If treated within days after the onset of brain damage, people are expected to make a complete recovery. However, one of the major challenges is making a proper diagnosis, since the symptoms of Korsakoff’s syndrome present similarly to other disorders.
Traumatic brain injury
A sudden blow to the head is likely to produce a traumatic brain injury (TBI). TBIs are the most common form of brain damage in younger people, with about 2.88 million Americans in 2014 with a TBI-related emergency department visit, hospitalization, or death. TBI most frequently results from automobile accidents, falls, high-contact sports, and due to occupational hazards such as in construction or military deployment, among many others. A concussion is a mild form of a TBI. Some of the common symptoms that people experience shortly after experiencing a TBI include double-vision, headache, dizziness, nausea and vomiting, and loss of consciousness, which are common symptoms in concussions as well.
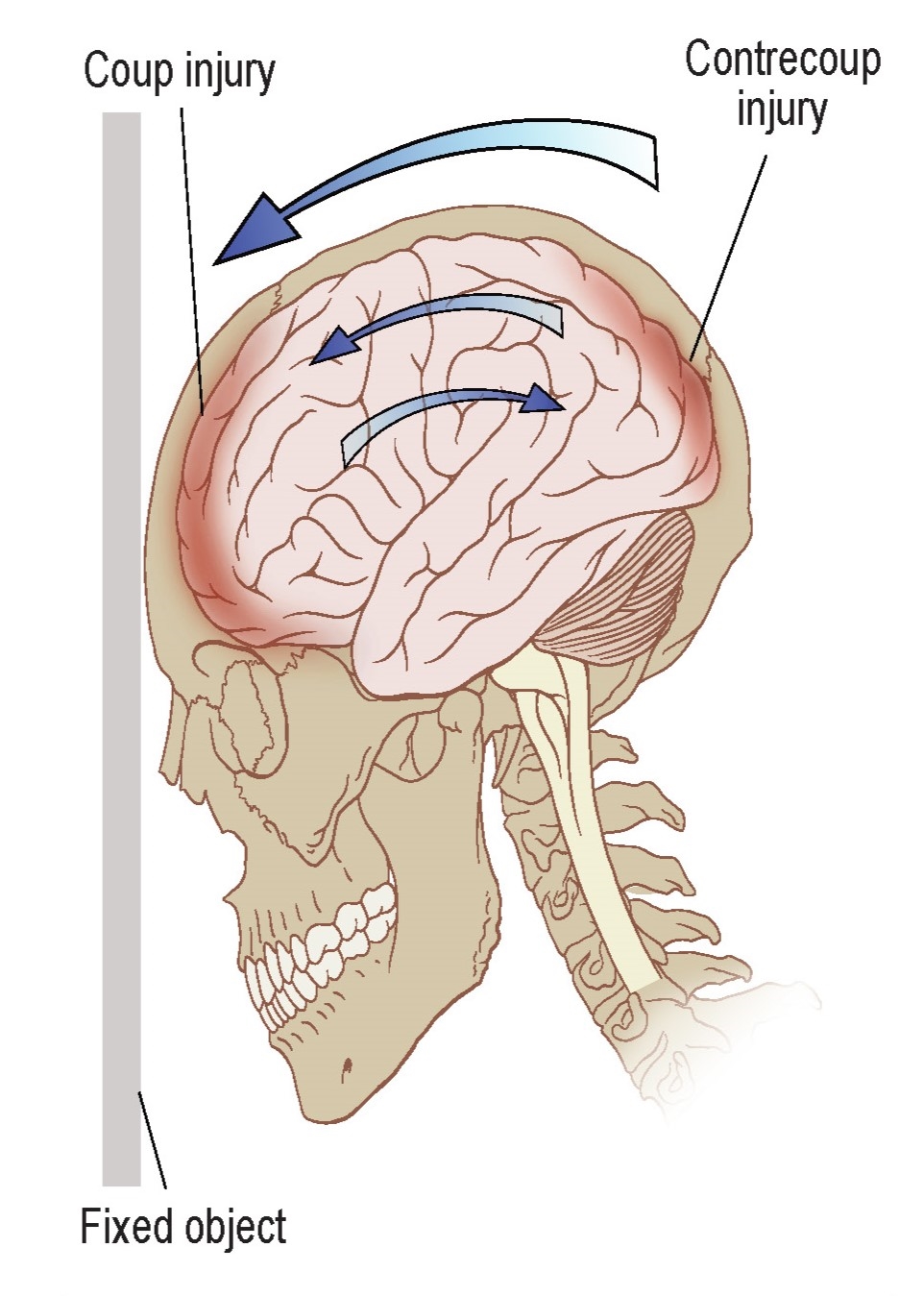
Usually in a traumatic brain injury, there are two simultaneous insults: the coup, which is the injury when the brain hits the inside of the skull closest to the external force. Shortly after, the contrecoup happens, which is the injury when the brain recoils backwards and hits the interior surface of the skull opposite of the cause of the insult. Following these injuries, there is a widespread inflammatory response that changes cellular activity in numerous complex ways, such as microglial activation, axonal stretching, and elevated levels of immune signaling molecules.
People with repeat head injuries may also experience some memory loss. It is suggested that the damage to the brain creates an abnormal cellular environment that interferes with the reverberation processes that are normally important for solidifying memories. Additionally, TBI decreases hippocampal neurogenesis, which is likely to impair the formation of declarative memories. One of the biggest difficulties is that TBI is very different from case to case, making development of new therapeutic strategies challenging. Currently, the best treatment for TBI is rest. There is still debate about the types of medications that can help best improve outcomes, psychostimulants and antidepressants being the most common classes of drugs used in TBI.